|
|
Molecular phylogeny |
| Systematics |
Molecular phylogeny |
Molecular phylogeny
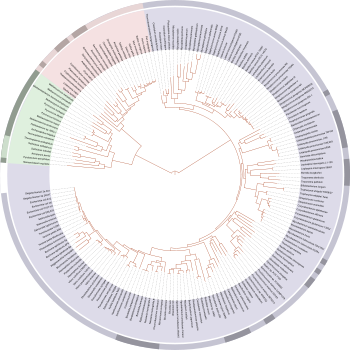
Tree of life according to molecular phylogeny (Wikipedia)
Molecular phylogeny, the sequencing and use of informational macromolecules such as DNA, RNA, and proteins for phylogenetic analyses in order to map out the evolutionary tree of life, has been, like cladistics, one of the great late 20th century revolutions in evolutionary understanding.
Like phenetics, molecular phylogeny uses computers to process large amounts of amounts of data for quantitative analysis, generating cladogram-like dichotomous branched trees, called phylograms. These contain more data than cladograms, because whereas cladograms represent branching order only, phylograms include the degree of evolutionary change as well.
Originally, molecular phylogeny used phenetic methods such as overall similarity, generating trees through less computation intense methods such as neigbour joining. With the phylogenetic revolution and the incorporatrion of cladistics, this approach was realised to be unsatisfactory, and molecular phylogeny now uses more sophisticated algorithms such as maximum parsimony, maximum likelihood, and bayesian inference. These methoids have been taken up in computational cladistics.
Because of their strong methodological similarity and cross-fertilisation, molecular phylogeny and computational cladistics have merged into a single discipline, called phylogenetics, and cladistic analyses now regularily use both molecular and morphological data for a combined (or total evidence) approach. But as cladistic morphology based and molecular phylogeny based trees very rarely agree in details of branching order (or topology to use the technical term), and often differ quite radically, the problem arises as to how to resolve this endemic phylogenetic incongruency between two otherwise very reliable phylogenetic systems.
The tendency among both cladists and molecular phylogenists has been to preference molecular-based tree topology, and therefore to force morphology-based cladograms to follow the molecular topology. As a result, paleontological trees tend to use morphology with fossils, but both morphology and molecules with recent taxa. The implication is that rampant and unbiquitous homoplasy means that morphology has a poor phylogenetic signal. Whether this is true or not is still debatable. In any case, molecular sequencing is assumed to give a more reliable signal both because of the larger and more easily quantifiable amount of data it provides, and because many DNA sequences are not affected by natural selection. Conversely, the fact that there are only four types of RNA/DNA necleotide bases makes the problem of homoplasy and long branch attraction artifacts even worse, hence the need for this to be corrected by appropriate statistical analysis, such as bayesian and maximum likelihood. The current emphasis is on incorporating both morphology and molecular data in a total evidence approach. MAK130414
page by MAK, edited RFVS111203, last modified MAK130414
