Molecular phylogeny
Molecular Phylogeny
Abstract
.
Introduction
Since the 1960s, when many of the breakthrough ideas of modern molecular biology were first published, the detailed composition 'sequences' – principally amino acid and nucleotide sequences) of biomolecules have become steadily better known. At first only protein sequences were available but later, as technology improved, DNA sequences became available as well. Homologous molecules were discovered in different organisms, and it soon became evident that the basic biomolecular framework of all living things is the same; an observation consistent with the very Darwinian notion that all life is, ultimately, monophyletic.
Molecular sequencing illuminates the evolutionary history of the molecules themselves and, consequently, that of their host organisms. By comparing homologous molecules from different organisms it is possible to establish their degree of similarity, thereby revealing a hierarchy of relationships: a phylogenetic tree. The continuing publication of sequences from diverse taxonomic groups has given rise to what David Penny (2002) describes as a small industry inferring evolutionary relationships.
"The general principle behind phylogenetic methods is to find a tree that minimizes sequence change. For example, if two species have a unique amino acid at a particular site and are joined in the tree, only one change (in their ancestor) is needed to explain this data. Conversely, an additional change would be required if the two species were not joined in the tree, making the other tree less likely to be the true tree. The two tree-building methods that are most often used with molecular sequence data are minimum evolution, such as neighbor joining, and maximum likelihood. These methods, and the Bayesian method, are flexible enough to include diverse information on the biological nature of molecular sequence change, such as rate variation among sites. A fourth method, maximum parsimony, is also widely used. Although the various methods are quite different from one another, they often result in the same phylogenetic tree. Reliability can be tested in different ways, with the bootstrap method being the most widely used. Phylogeneticists often use and compare several methods in a single study to evaluate the robustness of their results" (Hedges 2002, p. 839).
An assessment of molecular evidence, or of both molecular and morphological evidence, has often proved useful where morphological evidence alone has led to ambiguous results. Many different biomolecules are available for such analyses and this wealth of available characters is perhaps the greatest virtue of the biomolecular technique. Two illustrative examples are:
-
The Pogonophora are not well-known to most people, although one close relative, the giant red tube worms found living near hydrothermal vents along various deep sea trenches, are "almost famous." Pogonophorans long resided in an independent phylum and some Russian zoologists still maintain this interpretation), but molecular sequencing has confirmed ontological studies placing them in the Annelida, close to the polychaetes (Nielsen 2001, pp.
170-171).
-
Traditionally, zoologists have regarded molluscs and annelids as the closest relatives of arthropods. However, in 1997 that idea was challenged when Aguinaldo et al. (1997) proposed a clade they named Ecdysozoa, characterized by ecdysis, or moulting, under the influence of ecdysteroid hormones. The ecdysozoans are supposed to include arthropods, priapulids and nematodes. The ecdysozoan hypothesis has not been universally adopted, however. The chitin cuticle of arthropods may not be homologous with the collagen cuticle of nematodes (Adoutte et al. 2000). Additionally, Nielsen (2001, p. 119) mentions some critical 18S rDNA studies which have produced different phylogenies and concludes that the discrepancies will have to be resolved through further study.
Other advances in our understanding of the phylogeny of different groups – notably the protists and the angiosperms – also owe a great deal to molecular analyses.
Bayesian Method: The Bayesian method selects the tree that has the greatest probability that the tree is correct given under a specific model of substitution.
Bootstrap Technique: Randomly sized and positioned pieces of sequence from the same part of the molecule (from each organism) are sampled randomly, with replacement, and a new phylogenetic analysis is performed to produce a tree. This is repeated many times (normally 100). These bootstrap results are compared to the original approximated tree and each branch point is scored (agree or disagree). Scores around 50-60% are considered dubious; those up around 90% provide confidence that the predicted branch is accurate. Controversy arises when a branch is interpreted as meaning something, when in fact the score may say it is insignificant. [Adrian Walden, Vialactia Biosciences, pers. comm.]
Neutral Theory
The neutral theory of molecular evolution, proposed by Motoo Kimura in the 1960s, provides the theoretical underpinning for molecular phylogenetic research. The theory posits that the majority of mutations accumulated in any genome were neutral: 'neither beneficial nor injurious' in Darwin's words (Penny 2002). Thus mutations could accumulate continuously, providing the causal mechanism – the
'ticking' – of the clock.
"Both DNA and protein sequences are used for estimating phylogenetic relationships and times of divergence among taxa. Typically, DNA sequences are used for relatively recent events – for example, the human and chimpanzee split – when protein sequences are too conserved to be useful. Protein sequences are desirable for more ancient events – for example, human divergence from insects – when DNA sequences are usually too divergent to make accurate estimates on the basis of patterns of nucleotide substitutions. Unequal base or amino acid composition among the genomes of different species is common and makes sequence change more difficult to estimate. In addition, sequence length is a limiting factor, in that the average gene (coding) or protein sequence (~1,000 nucleotides, ~350 amino acids) is usually not long enough to yield a robust phylogeny or time estimate, and therefore many genes and proteins must be used" (Hedges 2002, p. 839).
Caveats
In many cases, phylogenies based on molecules are found to be robust and they are reinforced by subsequently discovered morphological or behavioral data. Yet, despite these successes, molecules have not proved as unambiguous as had been hoped. "Molecules, like morphologies, vary in their evolutionary rates and are subject to parallel and convergent evolution: and in consequence different molecules often suggest different phylogenies, just as do different morphological characters" (Arthur 1997, p. 53).
"The most useful single molecule has been ribosomal RNA (rRNA), which is homologous for all living organisms and, because it seems to keep evolving in secondary structure, its primary sequence is easier to use to reconstruct 'good' trees. Whether the trees are fully correct is another matter. There are at least two reasons why it is very difficult to resolve these deepest divergences. The first is that our models of the processes of mutation, and selection on individual sites of a macromolecule, predict saturation within 500 million years. Thus, we expect lower accuracy further back in time. The second difficulty is the lateral transfer of some genes. The best-established cases are the endosymbiotic origins of mitochondria and chloroplasts where their DNA sequences established an origin from bacteria. Both endosymbiotic and ectosymbiotic living arrangements are common in nature and therefore no unusual biological processes had to be invoked for their origin" Penny 2002).
The most commonly used rRNA subunit is 18S, comprising about 1800 base pairs, because it evolves slowly. Slow evolution is a prerequisite for probing very ancient phylogenetic events, to minimize the saturation problem. However, this same property makes the molecule unsuited for distinguishing events which occurred close together, perhaps during a rapid radiation. A possible example of this problem is found among annelids and molluscs; analyses which include many representatives of both phyla show a complete mix of the two groups. (Echinoderms, conversely, always appear as a monophyletic group, suggesting the modern forms represent a single lineage which diverged long ago, accumulating many unique mutations.)
"The horizontal transfer of genes is often difficult to confirm by phylogeny alone because the short length of typical proteins (~300 residues) usually precludes the construction of a robust tree, and different methods of detection do not always agree. Therefore,
'misplaced' species on a tree might be evidence of horizontal transfer or poor resolution" (Hedges 2002, p. 841).
For further reading, see Nielsen 2001, chapter 57.
Total Evidence Approaches
"Although the cladistic paradigm allows (some might say requires) simultaneous analysis of morphological and molecular data, this combination of evidence is rarely attempted. This is due, in part, to the sampling problems of molecular studies and the use of ground plans and single-character analysis in morphological work. … Although
'total' evidence is something of a misnomer, the concept – that all evidence currently available be used simultaneously – is hard to deny" (Wheeler 1997, p. 87).
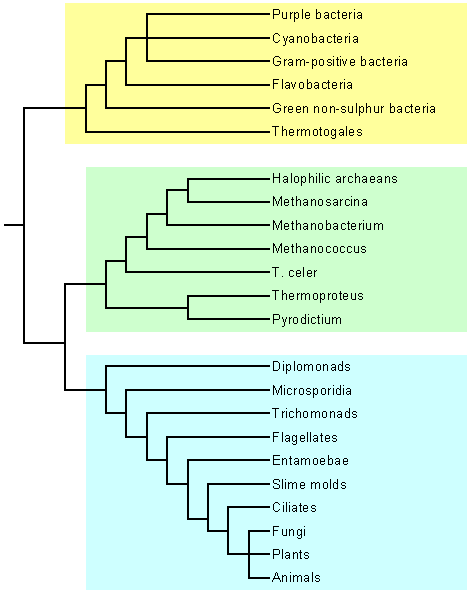 |
| Fig. 1: An early "Universal Tree of Life" deduced from ribosomal RNA (rRNA) data. The study upon which this figure was based did not resolve the branching of the three kingdoms most familiar to all of us: plants, Fungi and animals. Subsequent analyses, however, have revealed that the biochemistry of fungi (in particular, the synthesis of chitin) is most similar to animals. Thus, counter-intuitively, plants are likely to have diverged first, leaving fungi and animals as sister groups. (However, see Hedges 2002 for discussion of many inconclusive specifics.)
after Schopf 1999, p.105, fig. 4.2 |
The Universal Tree of Life
Although the basic molecular framework is the same for all life on earth, and thus very ancient, organisms are certainly different. More recently acquired physical (morphological) adaptations, adopted to enable their hosts to survive and prosper during the long course of evolution, are the basis for all but a few of the most recent revisions to our taxonomic view of the world. In the latter half of the twentieth century, biochemical studies have also come to compliment traditional comparative morphology. The morphological adaptations and biochemistry, too, are mirrored in the molecules.
The integration of many of these discoveries advanced in a quantum leap in the late 1970s. The standard view of the time, which had held sway for decades, was that the living world is fundamentally divided by the prokaryote-eukaryote dichotomy. This belief was challenged by Carl Woese and George Fox (Woese & Fox 1977) whose sequence analysis of
16S rRNA demonstrated that a division within the prokaryotes was at least as fundamental as that between prokaryotes and eukaryotes (fig.
1). "Analyses involving some unusual methanogenic 'bacteria' revealed surprising and unique species clusterings among prokaryotes. So deep was the split in the prokaryotes that Woese and Fox proposed in 1977 to call the methanogens and their relatives 'archaebacteria', a name which reflected their distinctness from the true bacteria or
'eubacteria' as well as contemporary preconceptions that these organisms might have thrived in the environmental conditions of a younger Earth" (Brown 2002).
"In 1990, Woese, Kandler and Wheelis formally proposed the replacement of the bipartite view of life with a new tripartite scheme based on three urkingdoms or domains; the Bacteria (formerly eubacteria), Archaea (formerly archaebacteria) and Eukarya (formerly eukaryotes although this term is still more often used)" (Brown
2002; but refer to Margulis et al. 2000 for an alternate view).
The Coalescence Method of Age Determination
If most mutations are neutral or almost neutral in their selective effects, they will tend to simply accumulate in their respective biomolecules over time. Provided the rate of this accumulation has not changed over time, then the rate of 'evolution' of a particular molecule should be approximately constant over time. The molecular clock hypothesis posits that a given biological molecule exhibits a relatively constant rate of change over time, irrespective of the taxonomic lineage within which it evolves (Zuckerkandl and Pauling
1962). For example, cytochrome c appears to have evolved at similar rates in vertebrates, in fungi and in plants. Although these organisms are phylogenetically diverse, their genes which code for cytochrome c exhibit convincingly similar rates of evolution.
If the rate of evolution of a particular molecule is nearly constant over time, we can use the degree of divergence between homologues in different taxa to estimate the time at which their evolutionary lineages separated. Even in those cases where some lineages have demonstrably different rates of evolution from other lineages (e.g. rodents vs. primates), provided we can identify homologous molecules which evolve at a constant rate, they can be used as 'clocks' to calculate the order in which lineages diverged. Furthermore, if we can calibrate the rate of change we observe, it may even be possible to estimate the age at which the lineages diverged from their nearest common ancestor.
Although a given biomolecule with the same function (such as cytochrome c) evolves at approximately the same rate in different taxonomic groups, this characteristic rate differs between different biomolecules. Some, such as the fibrinopeptides, change very rapidly; and others, such as the histones, change very slowly. Thus, for example, the rate for cytochrome c is considerably slower than the rate for hemoglobin.
Calibration
Before we can draw inferences from a molecular clock, we must calibrate its 'ticking rate.' Most often this is accomplished by pegging it against the known fossil record (keeping in mind that the first occurrence of a representative fossil is always a minimum estimate for the age of the lineage) though, occasionally, major geotectonic events, such as the isolation of a new landmass by rifting, can provide clues also. "Once the homologous gene A has been sequenced in, e.g., two species and the rate of evolution in this gene is known through prior calibration (let's say 2% per million years) then knowing the percent difference in the DNA sequence of gene A between these two species permits the calculation of the age of their last common ancestor. In this example, if species 1 and 2 differed by 10% in their DNA sequence of gene A, then the common ancestor of these two species would be expected to have lived around 2.5 mya. It would have taken these two lineages this long to both diverge at a rate of 2% per million years to accumulate 10% difference in gene A" (Mayr 2001, p. 198).
"[I]t can be shown that, on average, echinoderms and chordates are about 14% different in terms of their 18S rRNA sequences. [Cnidarian] differences are, on average, much greater – about 22% in each case. It seems, therefore, that as is expected from the comparative anatomy of these organisms, cnidarians left the line leading to chordates some time before the separation between echinoderms and chordates. The echinoderm-chordate split can be no younger than about
520 Ma because fossil echinoderms are found in Early Cambrian rocks. So, if it has taken ~500 Ma for a 14% difference to develop between echinoderms and chordates, the split between cnidarians and all other Metazoa – which resulted in a 22% difference – must have occurred some time prior to 520 Ma ago. If the rate of evolution was approximately constant over long periods of time, the split between Cnidaria and the other Metazoa could have been as early as 800 to 900 Ma before the present" (Runnegar 1992, p. 87).
Molecular dating, while not always in agreement with fossil evidence, offers an opportunity for timing events that are otherwise unobservable. For example, it was by the molecular clock method that the branching point between chimpanzee and man was shown to be as recent as 5-8 million years ago, rather than 14-16 million years, as had been previously generally accepted (Mayr 2001, p. 37; although latest fossil evidence hints that 5-8 Ma might be a slight underestimate). For the majority of evolutionary problems, the fossil evidence is either absent or inconclusive. With good calibrations, molecular data can greatly improve the constraints on timing estimates.
However, the molecular clock method must be applied with caution because molecular clocks are not nearly as constant as often believed. Not only do different molecules have different rates of change, but a particular molecule may vary its rate over time. These represent cases of mosaic evolution, in which evolutionary change occurs in a taxon at different rates for different structures, organs, or other components of the phenotype (Mayr 2001). This is a failure of the neutral assumption. If the mutations accumulating in a gene begin to express phenotypic effects which are subject to selection, then the rate of change can be effected. As noted above, multiple lines of evidence 'total' evidence) are preferable to dependence upon a single datum or technique.
(Also see Smith & Peterson 2002.)
In fact there is a probable echinoderm, Arkarua, described from the Ediacaran.
The recently described, 6-7 Ma Sahelanthropus tchadensis discovered at Toros-Menalla in Chad, is the oldest plausible human ancestor known to date. Not much younger, ~6 Ma, is Orrorin tugensis, discovered at Lukeino in Kenya. Together, the two fossil discoveries hint at a diverse and perhaps geographically widespread hominid ancestry, and an older divergence between men and apes than is indicated by most molecular studies. For the present, there is insufficient evidence to be sure.
Related Topics
Further Reading
-
Li, W-H (1997), Molecular Evolution, Sinauer Associates: 487 pp.
-
Liu, F-GR, MM Miyamoto, NP Freire, PQ Ong, MR Tennant, TS Young, & KF Gugel (2001), Molecular and Morphological Supertrees for Eutherian (Placental) Mammals Science 291: 1786-1789.
-
Shaffer, HB, P Meylan & ML McKnight (1997), Tests of turtle phylogeny: molecular, morphological, and paleontological approaches. Syst. Biol. 46: 235-268.
-
Shoshani, J & MC McKenna (1998), Higher taxonomic relationships among extant mammals based on morphology, with selected comparisons of results from molecular data. Mol. Phylog. & Evol. 9: 572-584.
Related Pages
Other Web Sites
References
Adoutte, A, G Balavoine, N Lartillot, O Lespinet, B Prud'homme & R de Rosa (2000), The new animal phylogeny: reliability and implications. Proc. Natl. Acad. Sci. (USA) 97: 4453-4456.
Aguinaldo, AMA, JM Turbeville, LS Lindford, MC Rivera, JR Garey, RA Raff, & JA Lake (1997), Evidence for a clade of nematodes, arthropods and other moulting animals. Nature 387: 489-493.
Arthur, W (1997), The Origin of Animal Body Plans: A Study in Evolutionary Developmental Biology. Cambridge Univ. Press, 338 pp.
Brown, James R. 2002 (in press): Universal Tree of Life. In Encyclopedia of Life Sciences. Nature Publishing Group, Macmillan.
Hedges, B (2002), The origin and evolution of model organisms. Nature Rev. Genet. 3: 838-849.
Penny, David 2002 (in press): Molecular Evolution: Introduction. In Encyclopedia of Life Sciences. Nature Publishing Group, Macmillan.
Margulis, L, MF Dolan & R Guerrero (2000), The chimeric eukaryote: origin of the nucleus from the karyomastigont in amitochondriate protists. Proc. Nat. Acad. Sci. (USA)
97: 6954-6959.
Martin, Andrew Peter 2002 (in press): Molecular Clocks. In Encyclopedia of Life Sciences. Nature Publishing Group, Macmillan.
Mayr E (2001), What Evolution Is. Basic Books.
Nielsen, C (2001), Animal Evolution: Interrelationships of the Living Phyla, [2nd ed.]. Oxford Univ. Press, 578 pp.
Runnegar, B (1992), Evolution of the earliest animals in JW Schopf [ed.], Major Events in the History of Life. Jones & Bartlett Publ. pp. 65-93.
Smith, AB & KJ Peterson (2002), Dating the time of origin of major clades: molecular clocks and the fossil record. An. Rev. Earth & Planet. Sci. 30: 65-88.
Tamas, Ivica; Klasson, Lisa; Canbäck, Björn; Näslund, A. Kristina; Eriksson, Ann-Sofie; Wernegreen, Jennifer J.; Sandström, Jonas P.; Moran, Nancy A.; Andersson, Siv G. E. 2002: 50 Million Years of Genomic Stasis in Endosymbiotic Bacteria. Science 296: 2376-2379.
Wheeler, WC (1997), Sampling, Groundplans, Total Evidence and the Systematics of Arthropods, in RA Fortey & RH Thomas [eds.], Arthropod Relationships. Systematics Assoc. Spec. Vol.
55: 87-96.
Woese, CR & GE Fox (1977), Phylogenetic structure of the prokaryotic domain: The primary kingdoms. Proc. Natl. Acad. Sci. USA 74: 5088-5090.
Zuckerkandl, E & L Pauling (1962), Molecular disease, evolution, and genic heterogeneity in M Kasha & B Pullman [eds], Horizons in Biochemistry. Academic Press, pp. 189-225.
page © Chris Clowes 2003
checked ATW030704, edited RFVS111203