The Bacteria
(Crown Group Life)
 Bacterial phylogenetics and systematics are areas that are fraught with controversy, confusion and very little concordance. To attempt to fit some sort of analysis of them within the confines of a single web-page, and without years of study to give oneself authority, would be the height of folly. With that in mind, feel free to read on, as we strive to raise ourselves to greater heights than ever before. If any of the arguments presented seem somewhat circular and self-contradictory, they probably are – you have been warned.
Bacterial phylogenetics and systematics are areas that are fraught with controversy, confusion and very little concordance. To attempt to fit some sort of analysis of them within the confines of a single web-page, and without years of study to give oneself authority, would be the height of folly. With that in mind, feel free to read on, as we strive to raise ourselves to greater heights than ever before. If any of the arguments presented seem somewhat circular and self-contradictory, they probably are – you have been warned.
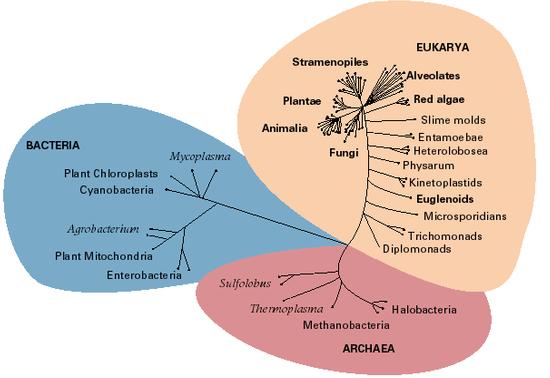
Undoubtedly the most influential work in modern higher-level prokaryote systematics was conducted by Carl Woese and associates in the 1970s and 1980s. This led to the much-popularised SSU rRNA tree in which life was divided into three "domains" separated from each other by long branches – the Eukaryota, the Archaebacteria, and the Eubacteria (later named by Woese as Eucarya, Archaea and Bacteria – Pace, 1997). The Archaea were the unexpected factor in this. They were found to possess a number of characters, particularly those relating to transcription of genes, in common with eukaryotes rather than other prokaryotes (Eubacteria), plus a few features entirely of their own. News headlines like ‘New third form of life discovered’ began appearing, and Woese’s redefinition of the term ‘Bacteria’ to include Eubacteria only, together with the unfamiliar extremophile nature of most of the cultured archaebacteria, lead to the establishment of the idea that Archaea were different in some fundamental way from Eubacteria.
Looked at from a more phylogenetic rather than a purely phenetic viewpoint, it becomes difficult to see what all the hyperbole is about. While Archaea have DNA-processing genes that resemble those of Eukarya, their metabolic genes are more like those of Eubacteria. Cavalier-Smith 2002). This only appears as a conflict if one assumes that all parts of the genome in all organisms are evolving at the same rate. This assumption is often made in molecular biology due to the influence of Kimura & Ohta’s (1974) Neutral Mutation Hypothesis, which suggests that the majority of genetic mutations are more or less selectively neutral in effect, so should happen randomly with respect to time. However, this theory only applies to mutations in non-coding parts of the genome, or other mutations that do not affect the resulting phenotype.
 When it comes to alterations in phenotype, different selective pressures on different parts of the genome and/or organism mean that evolution is not uniform for all characters of the organism – the principle known as ‘mosaic evolution’. Compare crocodiles and birds to their common reptilian ancestor – one is more distinct from the ancestor than the other, and within each, some features have changed more from the ancestor than others.
When it comes to alterations in phenotype, different selective pressures on different parts of the genome and/or organism mean that evolution is not uniform for all characters of the organism – the principle known as ‘mosaic evolution’. Compare crocodiles and birds to their common reptilian ancestor – one is more distinct from the ancestor than the other, and within each, some features have changed more from the ancestor than others.
Under this principle, the supposedly ‘inexplicable’ combination of characters possessed by Archaea is entirely explicable. Some of the features shared with one domain will represent plesiomorphies that have been lost in the remaining domain, while features shared with one or the other domain may be apomorphies of a larger clade.
To make any sense of this requires us to establish which domains are more closely related, and which is the most basalmost domain. This is where the real fun and frustration begins. The rRNA tree is, like all phylogenetic trees when they are first calculated, un-rooted. Normally the position of the root of a tree is established by inclusion of an outgroup, a taxon that is definitely known to be outside the group of interest. Unfortunately, somewhat by definition, no suitable outgroup exists for the totality of life. Obviously, a more inventive approach was needed.
 The approach used was to select genes that had duplicated before the Last Universal Common Ancestor of modern life (referred to by the catchy acronym ‘LUCA’). The trees of these genes should be able to be used to root each other and indicate the point where LUCA was to be found. The first two gene pairs used by independent researchers in 1989 were elongation factors (EF-Tu vs. EF-G) and catalytic vs. regulatory subunits of eubacterial F-ATPases with V- or V-like-ATPases of Eukarya and Archaea. Both these studies found the root to be on the branch separating Eubacteria from the other two domains. Philippe & Forterre (1999). Studies using other genes also found this pattern, and it became accepted as the standard view.
The approach used was to select genes that had duplicated before the Last Universal Common Ancestor of modern life (referred to by the catchy acronym ‘LUCA’). The trees of these genes should be able to be used to root each other and indicate the point where LUCA was to be found. The first two gene pairs used by independent researchers in 1989 were elongation factors (EF-Tu vs. EF-G) and catalytic vs. regulatory subunits of eubacterial F-ATPases with V- or V-like-ATPases of Eukarya and Archaea. Both these studies found the root to be on the branch separating Eubacteria from the other two domains. Philippe & Forterre (1999). Studies using other genes also found this pattern, and it became accepted as the standard view.
This picture of the evolution of life sat well with the supposed greater complexity of the DNA-processing systems in Eukarya and Archaea than in Eubacteria. Like all popular pictures, though, critics soon materialised to complain about it. Further gene studies failed to always retrieve the same branching order, and often didn't even recognise monophyly for the separate domains. Philippe & Forterre (1999); Cavalier-Smith (2002). Also, many of the genes used appeared to be mutation-saturated at the level used, so that the points of intersection of the paralogous trees were potentially the result of long-branch attraction. Philippe & Forterre (1999). For various reasons, most researchers in bacterial systematics continue to use rRNA trees exclusively, despite suggestions they may be unreliable (see below) and increased recognition in systematics of other organisms that phylogenetic evidence should be drawn from as many sources as possible. Division into three domains, with Eubacteria sister to Archaea + Eukarya, remains the norm, though a few alternative suggestions will be examined here.
The suggestion has been made that the common ancestor of all three domains was not yet a properly developed, integrated cell, but a ‘progenote.’ Woese (2002). Cell design was held to be shaped largely by rampant lateral gene transfer, with genetic components functioning as interchangeable modular units. Eventually, a ‘Darwinian Threshold’ was passed where genetic components of individual cells became integrated enough that lateral gene transfer was no longer able to occur enough to blur genealogical lines, and standard vertical descent became predominant. This threshold was passed separately in each of the three domains. The supposed sister status of Eukarya and Archaea is actually an artefact of analysis resulting from Eubacteria crossing the threshold earlier than the other two domains.
Support for this concept supposedly came from the wide divergence between the three domains, with completely different translation systems in Eukarya + Archaea vs. Eubacteria, plus the lack of phylogenetic resolution between domains and at the base of domains in trees for many genes. Translation systems were thought to have evolved independently in the two branches, thus removing the need to explain how one system replaced another. Multiple gene trees for Eubacteria show concordance at more recent nodes, but lower resolution at older nodes, potentially compatible with a ‘Darwinian Threshold.’ Creevey et al. 2004).
On the whole, though, this theory makes little sense. That LUCA lacked a translation system is not possible – it must have possessed one to have functioned as an organism. Characters such as the genetic code remain reasonably constant between domains, which would not be expected if it was independently derived in them. Therefore, a separate origin for the eukaryal and bacterial translation systems does not remove the need to explain the change of translation system – instead, we have to explain the replacement of the ancestral system by each of the derived systems. Also, as noted before, Archaea actually share many features with Eubacteria rather than Eukarya, and the differences are not as completely all-encompassing as often thought. The existence of a ‘Darwinian Threshold’ seems similarly tenuous – if lateral gene transfer was common in the past, there seems to be little reason why it should not still be so. The reasonable resolution in recent branches of gene trees argues against this – if anything, one would expect gene transfer to be more common between closely related organisms than distantly related ones, as there would be less chance that the newly-acquired genes would overly disrupt the genome of the recipient organism. It seems much more likely that the lack of resolution at more ancient levels is due as much to time eroding phylogenetic signal combined with rapid radiation of basal branches, as much as lateral gene transfer obscuring it. After all, Neoaves (the clade containing most modern birds) is also almost completely unresolved as to basal relationships, but no-one is suggesting lateral gene transfer between birds as the cause.
Philippe & Forterre (1999) suggested that Eukarya might be basal, with prokaryotes derived from eukaryotic ancestors by ‘genetic streamlining.' This suggestion was based on gene trees of slowly evolving positions of elongation factors. It was felt that this rooting ‘would best explain the presence of many more eubacterial-like genes than eukaryotic-like ones in completely sequenced archaebacterial genomes.' But, as explained before, there is no problem with this fact even if Archaea are sister to Eukarya. Archaea would then have simply retained mostly plesiomorphic features that have been lost in their sister group. A basal position for eukaryotes is also at odds with the fossil record. The earliest unequivocal eukaryotes are from the Late Proterozoic, about 850 My ago, though more doubtful examples are known from 1200 My ago. Either date is considerably younger than the earliest Eubacteria, which had appeared by 3.4 Gy ago at the latest (Cavalier-Smith, 2002).
 Archaea are often thought of as paraphyletic with regard to one or both of the other domains, with LUCA assumed to be archaebacterial in nature. Paraphyly with regard to Eubacteria, however, seems unlikely in light of the aforementioned greater complexity of DNA-processing systems in Archaea + Eukarya than in Eubacteria, probably due to DNA in the former group usually being contained by histones rather than DNA topoisomerases in Eubacteria (the former requiring more energy to disassociate than the latter – Cavalier-Smith, 2002). That these systems have not been ‘genetically streamlined’ in Eubacteria is supported by the fact that Eukarya and Archaea which lack or have reduced histones, such as Crenarchaeota and Dinoflagellata, retain the advanced processing systems rather than developing more eubacterial-like ones. Cavalier-Smith (2002).
Archaea are often thought of as paraphyletic with regard to one or both of the other domains, with LUCA assumed to be archaebacterial in nature. Paraphyly with regard to Eubacteria, however, seems unlikely in light of the aforementioned greater complexity of DNA-processing systems in Archaea + Eukarya than in Eubacteria, probably due to DNA in the former group usually being contained by histones rather than DNA topoisomerases in Eubacteria (the former requiring more energy to disassociate than the latter – Cavalier-Smith, 2002). That these systems have not been ‘genetically streamlined’ in Eubacteria is supported by the fact that Eukarya and Archaea which lack or have reduced histones, such as Crenarchaeota and Dinoflagellata, retain the advanced processing systems rather than developing more eubacterial-like ones. Cavalier-Smith (2002).
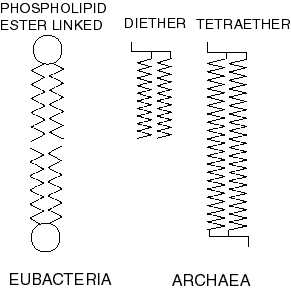
Paraphyly of Archaea with regard to Eukarya often appears in gene trees, but if Eubacteria is basal to Archaea + Eukarya, there is quite strong ‘morphological’ [1] evidence against it. Archaea possess a cell membrane composed of prenyl ether lipids, as opposed to acyl ester lipids in Eubacteria and Eukarya. Cell membrane characters are evolutionarily extremely stable, and this makes it much more likely that Archaea are a monophyletic sister-group to Eukarya. Cavalier-Smith (2002).
Also worthy of consideration is the suggestion that Eubacteria is actually paraphyletic with regard to Archaea + Eukarya. Cavalier-Smith (2002). Prokaryotes can be divided into two groups on the basis of cell membrane structure. The Monodermata or Unibacteria, containing Archaea and mostly Gram-positive Eubacteria, possess a single cell membrane. Didermata or Negibacteria, containing mostly Gram-negative Eubacteria, have a double membrane – the inner cytoplasmic membrane, and the more porous outer membrane. Cavalier-Smith made the argument that Didermata must be ancestral as loss of the outer membrane by hypertrophy of the murein wall between membranes was more probable than gain of a new membrane. While this theory is mechanistically plausible, the problem in evaluating phylogenies with mechanistic models is that Life has often proven to be more ingenious than researchers in coming up with pathways by which evolution may occur.
For now, I cravenly cower to the popular vote, and organise this page with the basalmost division on life between Eubacteria and Archaea + Eukarya. The tree for Archaea is taken from Cavalier-Smith (2002); see under Eubacteria for the rationale for the tree used for that domain. Names and information for divisions in Archaea are taken from Cavalier-Smith 2002), while names for Eubacteria are mostly taken from Garrity & Holt 2001), with some names taken from Cavalier-Smith (2002) for clades not recognised or named in the former source.
A few comments need to be made on the use of names for taxa. I have consistently used the name ‘Eubacteria’ instead of the recent (Woese et al., 1990) restriction of the name ‘Bacteria’ to this taxon only, despite the popularity of the latter usage. Archaea were previously universally regarded as bacteria, and terms such as ‘bacteriology’ and ‘bacterial’ are still often used to cover both Eubacteria and Archaea. The redefinition of ‘Bacteria’ was unnecessary as the name ‘Eubacteria’ is well-recognised, and doesn't have the same potential for double meaning.
The name ‘Archaebacteria’ was altered to ‘Archaea’ at the same time, to lose the implied connection to Bacteria. This also appears to be an unnecessary name-change. Names should not be changed merely because they are felt to be unsuitable for some reason – not only is it potentially confusing, but unsuitability is often (as in this case) a subjective matter that different researchers may disagree on. Despite the priority of Archaebacteria, the name Archaea has become more commonly used, and at least doesn't have the same potential for confusion as ‘Bacteria.' I therefore cave to popular pressure once more, and accept the name ‘Archaea’.
I have no pressing reason for using the name ‘Eukarya’ rather than ‘Eucarya’ or 'Eukaryota', other than personal preference.
© 2005 by Christopher Taylor. CT050119
Our tentative cladogram of the bacteria may be found, oddly enough, on the Cladogram page. Not to belabor the obvious, but the bacteria have been around a lot longer than anything else. Living species tend to be at the tail end of very long evolutionary chains; and, with rare exceptions, our knowledge is limited to living species. Consequently, there are pockets of diversity everywhere in the bacteria that don't seem to be very closely related to anything else. This makes it unreasonably difficult to summarize bacterial diversity. For the moment, we will have to make do with only the largest and most conspicuous groups
LUCA
├─
Eubacteria
│ ├─Actinobacteria
│ └─┬─Firmicutes
│ └─Didermata
│ ├─Cyanobacteria
│ └─┬─Sphingobacteria
│ └─Proteobacteria
└─Neomura
├─Archaea
└─
Eukarya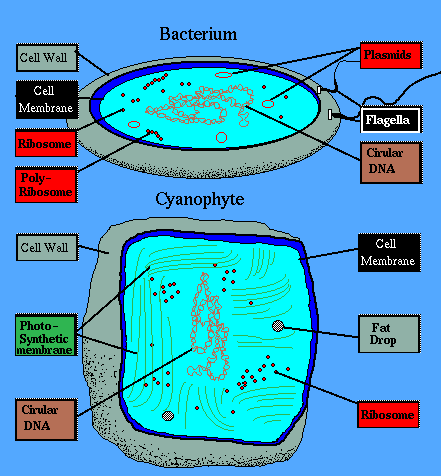 See Eubacteria: Cell wall of peptidoglycan or murein); cell membrane of acyl ester lipids. Flagellar shaft (if present) composed of flagellin. DNA replicative sliding clamp part of a type C DNA polymerase holoenzyme. Four RNA polymerase holoenzyme subunits. Hsp60 chaperonins with sevenfold symmetry and co-chaperonin Hsp10. CCA 3’- terminus of tRNA encoded by the gene. Protein synthesis initiated by N-formyl methionine. The main DNA helicase used in DNA replication is DnaB.
See Eubacteria: Cell wall of peptidoglycan or murein); cell membrane of acyl ester lipids. Flagellar shaft (if present) composed of flagellin. DNA replicative sliding clamp part of a type C DNA polymerase holoenzyme. Four RNA polymerase holoenzyme subunits. Hsp60 chaperonins with sevenfold symmetry and co-chaperonin Hsp10. CCA 3’- terminus of tRNA encoded by the gene. Protein synthesis initiated by N-formyl methionine. The main DNA helicase used in DNA replication is DnaB.
Relationships within Eubacteria are extremely uncertain at almost all levels of divergence, and many of those that have been suggested make little obvious sense. A number of factors have resulted in this situation – one is the general reliance on rRNA trees to the exclusion of other data sources. rRNA trees have been shown in recent years to be sensitive to variations in evolutionary rates in eukaryotes, leading to such errors as the placing of Microsporidia low down in the eukaryote tree, instead of in or near the Fungi. rRNA trees also show low resolution between most branches at high levels.
The other major issue with tree construction which has received a lot of attention is lateral or horizontal gene transfer (LGT), the direct transfer of genes from one species to another. The occurrence of LGT in prokaryotes between unrelated species is undoubted – however, opinions differ as to just how prominent it is. Some regard its occurrence as minimal (e.g. Cavalier-Smith, 2002), others feel that LGT may be so common as to render the construction of an organismal phylogeny for prokaryotes effectively impossible. This page tends away from the latter view, of course – if for no other reason than that otherwise we might as well give up and go home. Cases of LGT might even be potentially used as characters to support clades.
I have rather arbitrarily selected the rRNA tree in Miroshnichenko et al. (2003) to represent the general trend of current eubacterial phylogeny:
LUCA
├─Neomura
└─┬─Aquificae
└─┬─Thermotogae
└─┬─Dictyoglomus
└─┬─┬─Firmicutes
│ └─Cyanobacteria
└─┬─Actinobacteria
└─┬─┬─Caldithrix
│ └─┬─Deferribacteres
│ └─┬─Nitrospina
│ └─Thermodesulfobacterium
└─┬─┬─Sphingobacteria
│ └─┬─Spirochaetes
│ └─┬─Fusobacteria
│ └─Proteobacteria
└─┬─┬─Acidobacteria
│ └─Nitrospirae
└─┬─Eobacteria
└─Planctobacteria
Compare this to the tree in Cavalier-Smith (2002), constructed using mostly ‘morphological’ or physiological characters:
LUCA
├─Eobacteria
└─┬─┬─Cyanobacteria
│ └─┬─┬─Firmicutes
│ │ └─Thermotogae
│ ╘═╤═Actinobacteria (paraphyletic)
│ └─Neomura
└─┬─Spirochaetes
└─┬─Sphingobacteria
└─┬─Planctobacteria
└─Proteobacteria
As different as these two trees are, there are some similarities. Most notably, if both trees are unrooted, the Gram-positive bacteria (Actinobacteria, Firmicutes and Thermotogae) are close to Neomura corresponding to the Monodermata); the Didermata are mostly further away. The relationships within Didermata are more contradictory, but seem poorly supported in both papers (though neither paper actually gives any real measure of support). The exception is Cyanobacteria, which are closest to Monodermata in both trees.
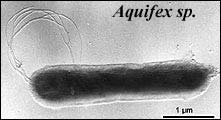 The differences in positions of the Aquificae and Thermotogae are the most significant differences between the trees. Aquifex has been widely accepted as the basalmost eubacterium due to its position in rRNA trees. However, Aquifex has a double membrane, suggesting a position within Didermata. Some protein trees place it within the ε-proteobacteria, and this was the position accepted by Cavalier-Smith. As placing Aquifex in its position on the rRNA tree implies multiple gains or losses of the outer membrane, I here tentatively accept the Cavalier-Smith position.
The differences in positions of the Aquificae and Thermotogae are the most significant differences between the trees. Aquifex has been widely accepted as the basalmost eubacterium due to its position in rRNA trees. However, Aquifex has a double membrane, suggesting a position within Didermata. Some protein trees place it within the ε-proteobacteria, and this was the position accepted by Cavalier-Smith. As placing Aquifex in its position on the rRNA tree implies multiple gains or losses of the outer membrane, I here tentatively accept the Cavalier-Smith position.
Thermotogae is the second-most basal major branch in rRNA trees, but is grouped with Firmicutes on many protein trees, by comparison of indels, and by the gene-content tree. As such, I accept the latter position on this page. The differences in position of the Thermotogae and Aquificae are probably due to long-branch attraction in the rRNA tree, and a high proportion of G+C in the genomes of these two taxa and Archaea.
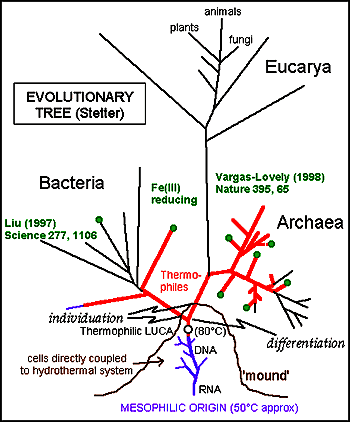
 The reposition of these two taxa has significant implications for one of the conclusions drawn from the rRNA tree of life – the supposed hyperthermophilic nature of Luca. This theory was supported by two of three domains having hyperthermophiles as basalmost members. With the eubacterial tree shown here, Archaea is the only domain that is still potentially basally hyperthermophilic, and a mesophilic Luca seems more likely. A hyperthermophilic origin of life, while thought to be consistent with widespread conditions on the young, newly-formed earth, is not consistent with the reduced stability of RNA at high temperatures.
The reposition of these two taxa has significant implications for one of the conclusions drawn from the rRNA tree of life – the supposed hyperthermophilic nature of Luca. This theory was supported by two of three domains having hyperthermophiles as basalmost members. With the eubacterial tree shown here, Archaea is the only domain that is still potentially basally hyperthermophilic, and a mesophilic Luca seems more likely. A hyperthermophilic origin of life, while thought to be consistent with widespread conditions on the young, newly-formed earth, is not consistent with the reduced stability of RNA at high temperatures.
In the tree used here, the order of branches between the base and Cyanobacteria is based on the Cavalier-Smith tree, while relationships within the Didermata exclusive of Cyanobacteria are, for now, based on the more familiar rRNA tree in light of their greater uncertainty. Most of the taxa are based on clusters in gene trees, and may be lacking in morphological apomorphies.
Actinobacteria are Gram-positive and almost exclusively aerobic. Their DNA is biased toward high G+C content. Actinobacteria contain 20S proteasomes. Such proteasomes are otherwise known only from Archaea and eukaryotes. Nagy et al. (1998). Often with snapping division or branching filaments; spores if present usually exospores. Filamentous members of this clade are often referred to as ‘fungi’. Actinomyces, Streptomyces, Mycobacterium, Propionibacterium, Corynebacterium, Nocardia, Micrococcus.
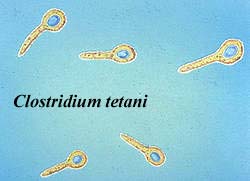
Mostly with thick rigid murein walls containing teichoic acids and lipoteichoic acids; often forming endospores. Clostridium, Bacillus, Lactobacillus, Streptococcus, Staphylococcus. In contrast, one subclade, the Mollicutes, has lost the cell wall, and is mostly intracellular parasites or symbionts Mycoplasma). The name ‘Firmicutes’ was originally coined to include all Gram-positive bacteria (including Actinobacteria and Togobacteria) and excluding Mollicutes, so its restriction to a subsection of its original content is somewhat unfortunate. It seems to have been accepted, however, so tough.
As mentioned above, these are mostly Gram-negative Eubacteria have a double membrane – the inner cytoplasmic membrane, and the more porous outer membrane.
The blue-green ‘algae’, probably the most familiar bacterial clade, and one of the few to be recognised before the advent of molecular data (the other was the Spirochaetes). Characterised by oxygenic photosynthesis with chlorophyll a. Flagella absent. A single genus, Gloeobacter, is recognisably basal to all others in lacking thylakoids. The clade Phycobacteria contains all other cyanobacteria, and has the chlorophyll contained in thylakoids. Phycobacteria have traditionally been divided into five orders on the basis of morphological colony characters. Chloroplasts are derived from Phycobacteria, though from which subclade is still unknown.
Cytoplasmic membrane with sphingolipids; outer membrane with lipopolysaccharide; flagella absent.
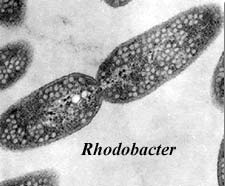 The largest bacterial clade – well-recognised by molecular data, but short on morphological synapomorphies. Large insertion in RNA polymerase and DnaK. While the genus Proteus is included within Proteobacteria, the division is not named after the gen us. Instead, both are named after the Greek shape-changing god Proteus – in the case of Proteobacteria, to reflect the wide range of morphologies covered by the clade. Includes photosynthetic purple sulphur (e.g. Rhodocyclus) and non-sulphur e.g. Rhodobacter) bacteria, intracellular parasites (e.g. Rickettsia), colonial formers of fruiting bodies (Myxococcales), and a wide range of heterotrophs, such as probably the most well-known bacterium of all, Escherichia coli. Divided by molecular data into five large clades, the α-, β-, γ-, δ-, and ε-proteobacteria. Examples – [Alphaproteobacteria] Rhodobacter, Rhizobium, Rickettsia; [Betaproteobacteria] Neisseria, Spirillum; [Gammaproteobacteria] Pseudomonas, Vibrio, Escherichia; [Deltaproteobacteria] Bdellovibrio, Myxococcus; [Epsilonproteobacteria] Helicobacter.
The largest bacterial clade – well-recognised by molecular data, but short on morphological synapomorphies. Large insertion in RNA polymerase and DnaK. While the genus Proteus is included within Proteobacteria, the division is not named after the gen us. Instead, both are named after the Greek shape-changing god Proteus – in the case of Proteobacteria, to reflect the wide range of morphologies covered by the clade. Includes photosynthetic purple sulphur (e.g. Rhodocyclus) and non-sulphur e.g. Rhodobacter) bacteria, intracellular parasites (e.g. Rickettsia), colonial formers of fruiting bodies (Myxococcales), and a wide range of heterotrophs, such as probably the most well-known bacterium of all, Escherichia coli. Divided by molecular data into five large clades, the α-, β-, γ-, δ-, and ε-proteobacteria. Examples – [Alphaproteobacteria] Rhodobacter, Rhizobium, Rickettsia; [Betaproteobacteria] Neisseria, Spirillum; [Gammaproteobacteria] Pseudomonas, Vibrio, Escherichia; [Deltaproteobacteria] Bdellovibrio, Myxococcus; [Epsilonproteobacteria] Helicobacter.
The Archaea, or Archaebacteria have cell membrane of prenyl ether lipids. Flagellar shaft of acid-insoluble glycoproteins related to pilin. DNA binding protein 10b. tRNA modifications, including archaeosine in D-loop and absence of queuine. Tiny large subunit ribosomal protein, LX. No Hsp90 chaperone. RNA polymerase A split into two proteins. Glutamate synthetase split into three proteins. Divided by rRNA trees into two major clades, Crenarchaeota and Euryarchaeota.
© 2005 by Christopher Taylor
last modified CT050119, edited RFVS111023