| Palaeos | Ascomycota | |
| Fungi | Pezizomycotina |
| Page Back | Unit Back | Fungi | Fungi Dendrogram | Fungi References | Glossary | Taxon Index |
| Page Next | Unit Next | Unit Home | Dendrogram | References |
ASCOMYCOTA |--Taphrinomycotina `--+--Saccharomycotina `--Pezizomycotina `--Pezizomycetes |--Hymenoascomycetes `--Loculoascomycetes |
Introduction |
 In our general section on Ascomycota, it was
our unfortunate duty to describe some aspects of the sexual reproductive cycle
of the Pezizomycotina. That discussion may be found on
another page. Having discharged this obligation, we decline to repeat the
performance here. It is still necessary for the reader to know the general outlines of
this stuff in order to make much sense of ascomycete phylogeny, so the reader is
referred to that earlier discussion -- with our sincere condolences.
In our general section on Ascomycota, it was
our unfortunate duty to describe some aspects of the sexual reproductive cycle
of the Pezizomycotina. That discussion may be found on
another page. Having discharged this obligation, we decline to repeat the
performance here. It is still necessary for the reader to know the general outlines of
this stuff in order to make much sense of ascomycete phylogeny, so the reader is
referred to that earlier discussion -- with our sincere condolences.
Truthfully, there is not all that much to say about Pezizomycotina as a taxonomic entity separate from Ascomycota. Euascomycetes, as Pezizomycotina was traditionally known, is a sort of inverse garbage taxon. As the old name implies, all ascomycetes which fit the classical concept of a well-behaved fungus of the general ascomycete type were called euascomycetes. Everyone else got put in a some special box. This is exactly the opposite of the way things tend to work in zoology. In zoology, well-behaved taxa are zealously packaged into innumerable, tightly-constrined systematic containers, while non-conforming animals tend to be lumped into broad garbage taxa. However, as we have mentioned elsewhere, the culture of mycology is unique. We will see more evidence of this below. In any event, the attention of mycology has been focused at other taxonomic levels, with Pezizomycotina simply serving as a general umbrella.
This is not to say that the taxon Pezizomycotina fails the test of monophyly. One of the reasons for the change of name was, presumably, to celebrate the taxon's graduation from a botanical type to a mycological clade. Nevertheless, although it is a phylogenetic entity, Pezizomycotina does not yet seem to have acquired a suitable phylogenetic definition, and its boundaries are a little unclear. As a result, although Pezizomycotina is evidently intended as a natural group of some sort, it is somewhat difficult to pin synapomorphies on it.
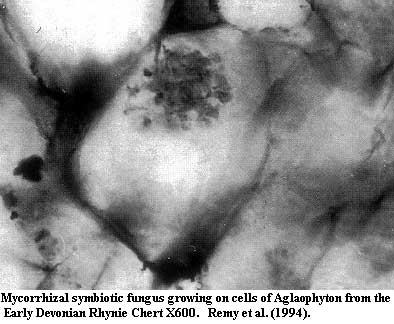 Pezizomycotines are first known in the fossil record from the Early
Devonian Rhynie Chert.
Certain remains from the Swedish Silurian
may also represent Pezizomycotina. Redecker
(2002). Claims for an ascomycete divergence date
anywhere from 600 to 1200 Mya (id.), based on "molecular clock"
models, currently lack substantiation.
It may be significant that the fungi which accompanied the first land plants in
the Ordovician were
neither ascomycetes nor basidiomycetes, but basal to both. Redecker
et al. (2000). This suggests that the evolution of these key groups,
or at least their widespread radiation, had not yet
occurred. Note also that both Ascomycota and Basidiomycota are
characterized by the absence of any type of cell motility, and by unique
reproductive characters which do not seem well designed for a marine
environment.
Pezizomycotines are first known in the fossil record from the Early
Devonian Rhynie Chert.
Certain remains from the Swedish Silurian
may also represent Pezizomycotina. Redecker
(2002). Claims for an ascomycete divergence date
anywhere from 600 to 1200 Mya (id.), based on "molecular clock"
models, currently lack substantiation.
It may be significant that the fungi which accompanied the first land plants in
the Ordovician were
neither ascomycetes nor basidiomycetes, but basal to both. Redecker
et al. (2000). This suggests that the evolution of these key groups,
or at least their widespread radiation, had not yet
occurred. Note also that both Ascomycota and Basidiomycota are
characterized by the absence of any type of cell motility, and by unique
reproductive characters which do not seem well designed for a marine
environment.
Nevertheless, the Rhynie Chert fossils probably represent (at least) Sordariomycetes and some sort of loculoascomycete -- i.e. highly derived pezizomycotines. Remy et al. (1994). Redecker (2002). Therefore, if the Pezizomycotina were not present in the Ordovician, they must have developed very rapidly in the following 50 My. The presence of possible lichenized forms with filamentous hyphae from c. 650 Mya are also evidence suggesting, but not requiring, the evolution of pezizomycotines by that date.
Pezizomycotina is notable for its association with plants, animals and protists. Redecker et al. (2000). One interesting example comes from recent studies attempting to characterize the mixed fungal/microbial/algal communities responsible for acid mine drainage. Baker et al. (2004) recently examined the phylogenetic composition of fungi in the mixed communities inhabiting this hostile environment: pH 1, 30-50° C., and containing about 0.3 M metal salts (mostly iron, but with significant amounts of, e.g., arsenic and copper). All of the fungi appeared to be pezizomycotines -- in particular, eurotiomycetes and dothidiomycetes. The fungi appear to play a structural role in the community, anchoring the biofilm to the underlying pyrite matrix. Baker et al. also speculate that they control the population structure of the community by selective grazing on the prokaryotic population.
This section, like the one on introns (below) has spawned a Pieces page on sugar chemistry and the fungal cell wall. As noted there, the cell wall is complex, with a poorly understood structure. The phylogenetic signal, particularly in the α(1→3)-glucan layer is obscure. However, the size and importance of α(1→4) side chains seems to increase over the course of pezizomycotine evolution, particularly in the Lecanoromycetes.
Standardized Dendrograms |
|
| Liu & Hall (2004) | Lutzoni et al. (2004) |
|
Pezizomycotina `--Pezizomycetes*
|--B--Chaetothyriales
| `--Dothidiomycetes*
| `--Arthoniomycetes
`--A--Eurotiomycetes
|--+--Sordariomycetes
| `--Leotiomycetes `--Lecanoromycetes A = Hymenoascomycetes B = Loculoascomycetes |
Pezizomycotina `--Pezizomycetes*
|--+--Sordariomycetes
| `--+--Dothidiomycetes
| `--Arthoniomycetes
`--Leotiomycetes*
|--Eurotiomycetes
| `--Chaetothyriales
`--Lecanoromycetes * = Paraphyletic |
We approach this section with great trepidation. The phylogeny of the Pezizomycotina is fraught with contradictions and inconsistency. Worse, we have reluctantly reached the conclusion that we're going to have to endorse a minority view. Specifically, we will reject the results of Prof. François Lutzoni and the AFTOL (Assembling the Fungal Tree of Life) Project and follow the phylogeny of Liu & Hall (2004). We have bent and stretched nomenclature to the breaking point in order to force both phylogenies into a head-to-head comparison, and into as close a correspondence as possible. However, as the table shows, the results of these studies are fundamentally dissonant.
The AFTOL approach combines truly massive amounts of rDNA and mtDNA data with sequence data from genes coding for RNA polymerase. Reeb et al. (2004); Lutzoni et al. (2004). Liu & Hall use only RNA polymerase amino acid sequence data, and a considerably smaller sample of taxa. However, it is likely that neither study uses a representative sample of euascomycete diversity, for the simple reason that only a small fraction of actual fungal diversity has been described. See, e.g., Schadt et al. (2003). In any case, this factor is slightly outweighed by the following considerations:
(1) Because of its history of generating bizarre phylogenies, we don't trust ribosomal DNA phylogenies, when there is anything else to rely on. In addition, they suffer from many of the same problems as mtDNA phylogenies. We don't trust mtDNA phylogenies under any circumstances, for reasons detailed elsewhere.
(2) There are particular reasons to be cautious when using fungal rDNA sequences.
(3) The AFTOL phylogeny has a slightly greater tendency to break up classical taxa into clumps that don't make any obvious biological sense; whereas
(4) Liu & Hall's phylogeny makes excellent biological sense. This, by a considerable margin, is the most important factor.
Ascomycete rDNA is, quite literally, pathological. At some point, probably about the time that Ascomycota and Basidiomycota diverged, the ascomycetes (and to a far lesser extent, the Basidiomycetes) were attacked by a sort of RNA parasite, the Group I introns. The background material on Group I introns refused to confine itself to a brief paragraph or two, so we have moved it to Pieces. This is unlikely to do you much good, since you are probably not on a first name basis with pAco.S788 and its relatives (Haugen et al., 2004). Accordingly, you will have to read the explanation anyway in order to make sense of what follows. Nonetheless, we derive a certain amount of wholly unmerited personal satisfaction from the superficial efficiency of referring you to this conveniently packaged discussion.
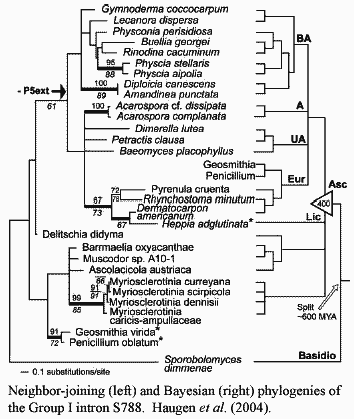 Unlike all other higher organisms, fungi contain a "vast array" of
both Group I and spliceosomal
introns in their rDNA. Haugen et al.
(2004). Spliceosomal introns are found only in Pezizomycotina,
and in fact, only in derived pezizomycotines. They are absent in the basal
Pezizomycetes. Bhattacharya
et al. (2000).
Unlike all other higher organisms, fungi contain a "vast array" of
both Group I and spliceosomal
introns in their rDNA. Haugen et al.
(2004). Spliceosomal introns are found only in Pezizomycotina,
and in fact, only in derived pezizomycotines. They are absent in the basal
Pezizomycetes. Bhattacharya
et al. (2000).
The Group I introns are somewhat more widely distributed. As of this writing (051105), 2696 Group I introns have been identified and sequenced. Of these, 1984 (74%) were isolated from eukaryotic nuclei, the remainder being from bacteria (3%) or organelles (23%). Of the roughly 2000 nuclear Group I introns, 67% are from Fungi generally and 61% from the Ascomycetes alone. Essentially all of the remaining Group I introns are from protists, with a handful known in land plants. No Group I introns have been isolated from metazoans. Cannone et al. (2002). Thus, unless the sampling has been extraordinarily poor, ascomycetes are peculiarly vulnerable to intron insertion. (Indeed, Haugen et al., 2004a report the bizarre case of an ascomycete with a Group I intron containing an internal spliceosomal intron).
It is not clear whether this chronic infestation is the result of a single event in the distant past, or whether entirely new infections have occurred at intervals. The S788 family of introns, for example, appears to have been vertically transmitted (with a few exceptions) since Precambrian times, since one, highly divergent, member of the family is found among the basidiomycetes. Haugen et al. (2004). On the other hand, a few Group I introns are known in all major fungal groups except the Microsporidia [1]. Cannone et al. (2002). At any rate, the ascomycetes have been infected and periodically cross-infected with these rDNA parasites for their entire history.
It is difficult to tell what effects this may have had on the underlying rDNA sequences. In all likelihood, there has been irregular selection against intron splicing sites, irregular insertions of partial intron copies or degraded spliceosomal intron sequences. The real point is that we just don't know what effects this pathology may have had; but the effects, whatever they may have been, are likely to have included irregular jumps and twists in the rDNA family tree.
Euascomycete rDNA is also unusual in another way. It has recently been discovered that the multiple copies of the genes coding for the 5S rRNA of ascomycetes are not identical. Not only may a single organism contain many different 5S "alleles," but it may also contain 5S pseudogenes and incomplete copies. Rooney & Ward (2005). For a number of reasons, we would not expect to see this kind of gross sequence heterogeneity in other rDNA species. However, we know to a certainty that other fungal rDNA repeats contain some sequence heterogeneity due to the variable presence and variable sequence of introns. So, how certain can we be that these rDNA phylogenetic probes are not also, to some degree, heterogeneous mixtures of independently evolving genes?
On the whole, we have reason to be particularly suspicious of rDNA phylogenies among the Ascomycota. Thus, the absence of rDNA data from the Liu & Hall study is likely to be a point in its favor.
 Each generation of scientists tends to believe, in its heart, that all
previous generations suffered from moderately severe brain damage.
Mycologists are, on the whole, less subject to this prejudice than most.
Yet,
some of this prejudice is also present in mycology. However, as cladistic and
molecular methods have matured, we are finding with astonishing frequency that
the Ancients weren't idiots after all. The astonishing thing is that they
were able to accomplish so much accurate phylogeny with nothing more than
intuition and gross morphology. Some key mistakes were made -- in
mycology, the unfortunate absorption with botanical concepts, and the inclusion
of the oomycetes among fungi, to name two. Nevertheless, previous
generations of systematists were, on the average, keener observers then we are today
because phyletic observation was essentially the only tool they had.
Consequently, absent convincing evidence to the contrary, we ought to prefer a
phylogeny which preserves the traditional taxa.
Each generation of scientists tends to believe, in its heart, that all
previous generations suffered from moderately severe brain damage.
Mycologists are, on the whole, less subject to this prejudice than most.
Yet,
some of this prejudice is also present in mycology. However, as cladistic and
molecular methods have matured, we are finding with astonishing frequency that
the Ancients weren't idiots after all. The astonishing thing is that they
were able to accomplish so much accurate phylogeny with nothing more than
intuition and gross morphology. Some key mistakes were made -- in
mycology, the unfortunate absorption with botanical concepts, and the inclusion
of the oomycetes among fungi, to name two. Nevertheless, previous
generations of systematists were, on the average, keener observers then we are today
because phyletic observation was essentially the only tool they had.
Consequently, absent convincing evidence to the contrary, we ought to prefer a
phylogeny which preserves the traditional taxa.
 Neither
the Lutzoni nor the Liu & Hall phylogenies invariably preserves the classical
taxa, although both make claims to classical antecedents. However, Liu
& Hall score slightly better. Both find that Pezizomycetes is
paraphyletic. Liu & Hall also take the entire Chaetothyriales out of
Eurotiomycetes and make it sister to Dothidiomycetes -- but so do a good many
classical mycologists. Finally, they slip Baeomyces,
a lichenized dothidiomycete, in with the Lecanoromycetes by relabelling the
group with the unlatinized term "Lichenized."
Neither
the Lutzoni nor the Liu & Hall phylogenies invariably preserves the classical
taxa, although both make claims to classical antecedents. However, Liu
& Hall score slightly better. Both find that Pezizomycetes is
paraphyletic. Liu & Hall also take the entire Chaetothyriales out of
Eurotiomycetes and make it sister to Dothidiomycetes -- but so do a good many
classical mycologists. Finally, they slip Baeomyces,
a lichenized dothidiomycete, in with the Lecanoromycetes by relabelling the
group with the unlatinized term "Lichenized."
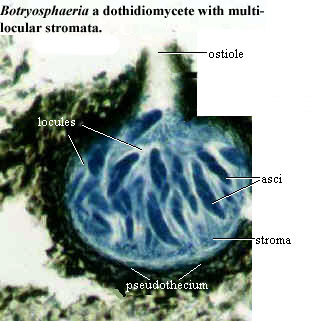
On the other hand, the AFTOL phylogenies tend to fragment both Dothidiomycetes and Leotiomycetes each into several parts. The Leotiomycetes are a vague lot, and even Liu & Hall insist only on the monophyly of one portion, the Heliotiales. However, the Dothidiomycetes are a reasonably distinct and specialized bunch. They include the Pleosporales, such as Pleospora, which was described previously. As in Pleospora, development is ascolocular and the asci are usually associated with pseudoparaphyses in a pseudothecium and are bitunicate. (Look, we told you that you were going to have to review the page on pezizomycotine reproduction. But did you heed this warning? Of course not!) We are uncomfortable breaking up this kind of taxon, absent more compelling reasons to do so.
Finally, a prior paper by the Lutzoni group, Reeb et al. (2004) introduces and circumscribes several novel large taxa which are carried forward into Lutzoni et al. (2004). It is hard to know what to make of these taxa without a more specialized knowledge of fungal anatomy than we yet possess. Yet, the circumscriptions these taxa are couched in phrases such as "ascomata immersed, sessile or pedunculate in form of apothecia (cryptolecanorine, lecanorine, rarely lecideine-immersed) or in form of perithecia." Reeb et al. (2004: 1055). This is an intimidating barrage of terminology. But the bottom line, we suspect, is that not much is excluded when so many possibilities are included, and the morphological criteria thus look suspiciously uninformative.
Here is where Liu & Hall sharply differ from Lutzoni et al. Putting together a nice molecular phylogeny is like winning at solitaire. To do either one consistently takes an understanding of the rules and a command of a large, but sharply delimited, set of data. But neither feat means anything by itself, because there is no real-world connection. To graduate from being an interesting, but pointless, expenditure of computing power, molecular phylogenetics has to make sense in terms of real-world biology. Liu & Hall do a spectacular job of showing why and how their phylogeny relates to biology. Lutzoni et al. don't. We do not say that the AFTOL group could not do so, but they have not done so yet.
Liu & Hall Phylogeny(rearranged to emphasize evolution of developmental characters) |
Pezizomycotina: unitunicate, apothecial `--Pezizomycetes: no change |--Hymenoascomycetes: no change | |--+--Leotiomycetes: no change | | `--Sordariomycetes: unitunicate, perithecial | |--Lecanoromycetes: no change | `--Eurotiomycetes: unitunicate, cleistothecial `--Loculoascomycetes: bitunicate, locular |--Dothidiomycetes: no change `--Chaetothyriales: same, pseudoparaphyses |
At this point we ought to summarize Liu & Hall's discussion of biological correspondences in some detail. However, their paper is a short one, and it can be found on the web at the PNAS web site -- to be precise, here. Thus, unlike many papers on the web, Liu & Hall (2004) is likely to be available indefinitely. We will therefore leave the reader to her own devices, except for the sections of the paper concerning the evolution and initial diversification of the Pezizomycotina.
Like Lutzoni et al., Liu & Hall find that Neolecta and yeasts (Saccharomycotina) are the sister groups of Pezizomycotina. As noted earlier, Neolecta has a basic form of ascus. This makes it likely that the euascomycete ascus is directly inherited from a Neolecta-like taphrinomycote. The yeasts therefore represent a side branch of development from ancestors who were multicellular and already possessed mycelial growth, specialized tissues, and even some of the basic euascomycete reproductive equipment. However, Neolecta lacks paraphyses, and the croziers do not develop until the ascus is formed. These characteristics probably represent the primitive state.
 Also
like the AFTOL group, Liu & Hall find that Pezizomycetes, the basal taxon of
Pezizomycotina, is paraphyletic, with Peziza the most basal member of the
series. Since Peziza and other basal pezizomycetes show
ascohymenial development (as opposed to development in a locule), an apothecium,
and unitunicate asci, these appear to constitute the primitive state within
Pezizomycotina. Thus the unitunicate ascohymenial growth form is
basal and we would expect it to have a parpaphyletic distribution. This is
exactly what Liu & Hall find.
Also
like the AFTOL group, Liu & Hall find that Pezizomycetes, the basal taxon of
Pezizomycotina, is paraphyletic, with Peziza the most basal member of the
series. Since Peziza and other basal pezizomycetes show
ascohymenial development (as opposed to development in a locule), an apothecium,
and unitunicate asci, these appear to constitute the primitive state within
Pezizomycotina. Thus the unitunicate ascohymenial growth form is
basal and we would expect it to have a parpaphyletic distribution. This is
exactly what Liu & Hall find.
However, for the same reasons, we would expect monophyletic distribution of the ascolocular, bitunicate mode of development. This is exactly where Liu & Hall's phylogeny differs from the AFTOL tree. In the AFTOL tree, ascolocular development is either independently developed three times, or developed twice and reversed once. Nearly anything is possible to evolution, given enough time; but this is not exactly a parsimonious solution. Liu & Hall's phylogeny, by contrast, requires monophyletic development of these highly specialized tissues. This happens mainly because, compared to the scheme of Lutzoni et al., the Chaetothyriales switch places with the Sordariomycetes. This switch creates three different molecular lineages within the ascolocular clade; and each of these molecular lineages corresponds to one of three identifiable, morphological variations on ascolocular development -- a really wonderful result.
The purpose of a phylogeny is to explain the evolution of organisms. With sufficiently sophisticated numerical methods, and a judicious selection of assumptions, even the most chaotic data can be rearranged to form some kind of statistically coherent pattern -- but that isn't the point of the exercise. Molecular methods merely maximize particular measures of internal statistical consistency. The idea is that such measures serve as numerical proxies for the life, death, reproduction and change of real organisms. However, there are no guarantees: that these measures are an adequate proxy, that the assumptions made are correct, or that the particular genes examined have evolved in a manner consistent with the assumptions and the statistical metric. A phylogeny is a hypothesis; and, in science, a hypothesis must be testable and should be tested. That can only be done by comparing a phylogeny with what we know about biology, chemistry, the fossil record -- whatever may be available.
Liu & Hall's phylogeny has passed one such test with a very high score. That's a good start.
 In addition to the usual molecular phylogenetic trees, the
full sequencing of several fungal genomes has permitted other types of
phylogenetic exploration. A particularly interesting series of experiments
was recently reported by Gasch
et al. (2004). These workers chose a number of short DNA
sequence motifs which have been identified as regulatory binding sites in the
yeast genome. Thus, for example, the sequence TCACGTG is a binding site
for centromere binding factor 1p ("Cbf1p"). In Saccharomyces
cerevisiae,
Cbf1p sites are important in the regulation of genes involved in methionine biosynthesis [2].
Gasch & Co. then located the orthologous genes in a number of other
ascomycete species and tested to see whether their upstream regulatory regions
contained the same binding site.
In addition to the usual molecular phylogenetic trees, the
full sequencing of several fungal genomes has permitted other types of
phylogenetic exploration. A particularly interesting series of experiments
was recently reported by Gasch
et al. (2004). These workers chose a number of short DNA
sequence motifs which have been identified as regulatory binding sites in the
yeast genome. Thus, for example, the sequence TCACGTG is a binding site
for centromere binding factor 1p ("Cbf1p"). In Saccharomyces
cerevisiae,
Cbf1p sites are important in the regulation of genes involved in methionine biosynthesis [2].
Gasch & Co. then located the orthologous genes in a number of other
ascomycete species and tested to see whether their upstream regulatory regions
contained the same binding site.
Not surprisingly, the results indicated that other Saccharomyces species and other closely related yeasts had substantially the same regulatory motifs in the same places. The phylogenetically interesting result was the rate at which this similarity fell away. Candida, a less closely related saccharomycotine, shared barely a third of these regulatory sites. The Pezizomycotina and Taphrinomycotina outgroup members shared only a very few regulators. The retained motifs corresponded to a small handful of very high level regulators at the top of extensive regulatory cascades [3].
These results fit a developing pattern with at least two important consequences for the way evolution works. First, it suggests that gene regulation -- at least in eukaryotes -- is hierarchical, rather than reticulated (net-like). Thus, evolution can take place by adding or tuning successively finer and more localized levels of control. This has the consequence of (a) creating a long-term trend toward higher complexity and (b) creating an environment which permits cellular specialization. By contrast, in a reticulated system, any change in one regulatory system will tend to affect all other systems. The probability of introducing a lethal side-effect goes up exponentially with complexity, so that the evolutionary potential of a reticulated system is limited. This is precisely the reason that other large, interacting systems, such as computer programs, must be built in a hierarchical, modular fashion.
Second, what the specific results of Gasch et al. (2004) also tell us is that, at least in fungi, this process is not overly conservative. That is, evolution doesn't simply continue to add more layers of control. Only the very highest level controllers are conserved in ascomycete evolution. Thus, the observed progressive evolution at low regulatory levels is eventually capable of re-engineering everything else in the organism, as localized changes incrementally affect higher levels of regulation. Ultimately, only the very highest levels of regulation are conserved.
In conceptual terms, Gasch et al. provide the beginnings of a precise answer to the noisy, but data-poor, debates about "micro-evolution" vs. "macro-evolution."
ATW051126. No rights reserved.
[1] Recall that Microsporidia have minimal genomes, probably due to their mode of infecting metazoan cells. Any unnecessary code would have been eliminated during the process of genome miniaturization.
[2] One problem with Gasch et al. (2004) is that the paper grossly oversimplifies the specificity of the DNA binding factors. In this example, Cbf1p binding is not only involved in methionine biosynthesis, but also in cysteine synthesis, as well as in completely unrelated activities such as DNA repair. Ferreiro et al. (2004); Kuras et al. (1997). Sure, a more complete description would make the paper five times longer; but, for an on-line journal like PLoS Biology, why is that a problem?
[3] Examples include the Mlu1 cell cycle box (MCB) and General Control Nondepressible Factor 4 (Gcn4p). MCB sits at the top of a cascade which promotes vegetative cell growth and nucleotide synthesis. Machado et al. (1997); Pilpel et al. (2001). Gcn4p may be even more general. It not only serves as a master switch to promote at least 12 different amino acid synthesis pathways, but also serves as a high-level regulator of glycolysis and other elements of vegetative metabolism. Hinnebusch & Natarajan (2002); Natarajan et al. (2001).
| Page Back | Page Top | Unit Home | Page Next |